254e Ab Initio Reaction Path Analysis of the Initial Hydrogen Abstraction from Organic Acids by Hydroxyl Radicals
Introduction
Carboxylic acids are important chemical constituents of the troposphere. They contribute to acid precipitation and can be detrimental to the environment and to human health. To elucidate the lifetime of these compounds and identify the major emission sources, the fate of these compounds needs to be well understood. Carboxylic acids are typically removed via photochemical oxidation reactions. The initial step in these reactions involves a hydrogen abstraction by a hydroxyl radical. This step is believed to be the rate limiting step of the oxidation reaction and to a large extent determines the product distribution of the reaction. Based on the thermodynamics of the hydrogen abstraction reaction, it has been proposed that the weaker C-H bonds, i.e. in the α-position of the carboxylic group, are most readily attacked.[1] However, experimental data seems to suggest that hydrogen abstraction occurs as well at the β-, γ- and d-position, and even further away from the –COOH group.[1,2,3,4] To better understand the kinetics of the oxidation of organic acids, we have performed high level CBS-QB3 quantum chemical calculations for hydrogen abstraction at the different positions in valeric acid, C4H9COOH, by a hydroxyl radical. Previous benchmark studies have shown that this computational procedure can predict reaction rate coefficients within a factor four of experimental data for hydrogen abstraction reactions. [5]
Results and discussion
Hydrogen abstraction by a hydroxyl radical at five different positions, i.e. at the -COOH group, at the α-, β- and γ-position and at the –CH3 group, was considered. The most stable radical is the α-radical, with an enthalpy of formation of -318 kJ/mol. For the β- and the γ-radical, enthalpies of formation of -295 and -397 kJ/mol, respectively, were calculated. Hydrogen abstraction at the CH3-group and at the COOH group are thermodynamically even less preferred. Based on thermodynamic arguments, the reaction is expected to proceed via the formation of the α-radical.
However, reaction rate coefficient calculations modify this picture. The transition state for hydrogen abstraction by a hydroxyl radical in the β-position was found to be 12.3 kJ/mol lower in energy than the transition state for the α-position, leading to a factor 140 difference in reaction rates at 298 K. The preference for the β-position is caused by the formation of a hydrogen bond in the transition state (Figure 1). The transition state is characterized by a 7-membered ring, where the OH radical forms a hydrogen bond with an oxygen of the carboxylic group. Due to ring strain, no hydrogen bond is formed in the transition state for the α-position. Additional calculations indicate that the formation of a hydrogen bond in the transition state lowers the activation energy by about 17 kJ/mol. Also for the transition state of the hydrogen abstraction at the γ-position, a hydrogen bond between the attacking OH radical and an oxygen of the carboxylic group was observed. However, the hydrogen bond is slightly weaker and the activation energy is slightly higher than for the β-position. In addition, the larger ring size leads to a lower pre-exponential factor and hence a lower reaction rate coefficient.
For each reaction pathway, a pre-reactive hydrogen bond complex was found along the reaction path, stabilized by about 20 kJ/mol. For hydrogen abstraction at the β- and γ-position the transition states were calculated to lie below the energy level of the reactants (Figure 1). The most stable complex was located on the reaction path for the hydrogen abstraction at the β-position.
Conclusions
High level CBS-QB3 quantum chemical calculations were performed to help elucidate the selectivity of the first step in the photochemical oxidation of organic acids. Activation energies were calculated for the abstraction of hydrogen atoms in valeric acid by a hydroxyl radical. Based on thermodynamic arguments, the α-abstraction pathway would seem to be preferred. However, detailed kinetic calculations reveal that abstraction of the β-hydrogen is the preferred mechanism. The preference for the b-hydrogen can be traced to the formation of a hydrogen bond in the transition state.
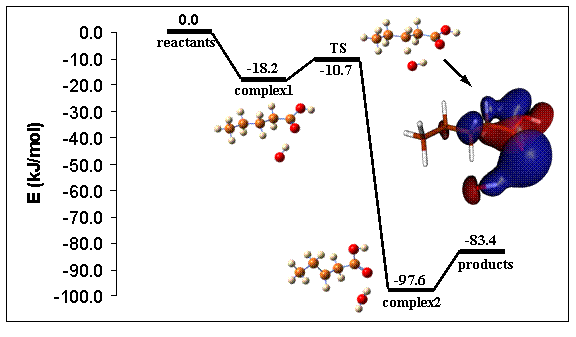
Figure 1. Potential energy profile for the hydrogen abstraction at β-position. The molecular orbital picture illustrates the hydrogen bond for the transition state.
References
[1] B.J. Finlayson-Pitts, J.N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments and Applications, Academic Press, San Diego, 2000.
[2] P.E.L. Williams, P.L. Silveston, R.R. Hudgins, Can. J. Chem. Eng. 1975, 53, 354-355.
[3] F. Jin, T. Moriya, H. Enomoto, Environ. Sci. Technol. 2003, 37, 3220-3231.
[4] H. Taniguchi, K. Fukui, S. Ohnishi, H. Hatano, H. Hasegawa, T. Maruyama, J. Phys. Chem. 1968, 72, 1926–1931.
[5] M. Saeys, M. Reyniers, V. Van Speybroeck, M. Waroquier, G. B. Marin, ChemPhysChem 2006, 7, 188-199.