616b The Effect of Rhodium Precursor on Carbon Monoxide Hydrogenation Catalysts
Introduction
During the 1980s there was a significant body of work concerning the production of oxygenates, from CO and hydrogen via a heterogeneous route [1, 2]. The optimum systems for such reactions were based on rhodium doped with various metals namely lithium, potassium, manganese, vanadium, zirconium and iron [3, 4, 5]. There has however been little work concerning the nature of the rhodium precursor and its effect on the activity and selectivity on the catalysts in question. Experimental
ĀIn this work two CO hydrogenation catalysts were prepared, the only difference being the nature of the rhodium precursor. The catalysts, prepared via incipient wetness, were of the following composition; 3 wt % Rh-Li(1/3)Mn(2/3)Zr(1/8)-silica where the numbers in brackets correspond to the atomic % w.r.t. Rh. The catalysts were synthesised using either RhCl3 or Rh acetate as the precursor.
The CO hydrogenation reactions were carried out in a Berty reactor at 50 bar pressure, 593 K with a CO:H2 ratio of 2:1, the set of conditions found to be optimum for the catalysts in question. The catalysts were subject to BET, XRD and temperature programmed reduction (TPR) prior to reaction and to temperature programmed oxidation (TPO) post reaction. Results and discussion
TPR of the fresh samples (Figures 1 and 2) showed that the chloride prepared catalyst was reduced in two stages with hydrogen uptake observed at both 154 oC and 191 oC. The acetate prepared catalyst was reduced in a similar fashion with peaks corresponding to hydrogen uptake at 127 and 181 oC.
XRD patterns taken for both catalysts showed that while the chloride prepared catalyst remained amorphous after calcination, the beginnings of the pattern for Rh2O3 (2 theta 32.7 o, 34.9 o and 48.7 o) was clearly observed forĀ the acetate prepared catalyst.
XRD patterns taken after a 450 oC reduction showed the emergence of peaks corresponding to metallic Rh for the chloride prepared catalyst (2 theta 41.0 o, 47.8 o and 70.0 o). This was in contrast to the acetate system where the beginnings of solid solution formation between the Rh and the other additives was seen (2 theta 34.0 o, 37.0 o, 41.5 o, 45.5 o and 55.0 o).Ā Studies on the hydrogenation of CO for the two different catalysts revealed significant differences in the activity/selectivity profiles. The conversion of the chloride prepared catalyst was found to be in the region of 6 % at steady state whereas that of the acetate prepared catalyst was 3 %. Not only was the acetate prepared catalyst less active, it was also less selective towards oxygenates (45% compared to 70%).
The results of the XRD and TPR experiments go some way to explaining the significant difference in activity between the two catalysts. An increase in selectivity towards oxygenates would be observed if the amount of delta positive Rh atoms on the surface of the catalyst was increased. Such species would decrease the likelihood of CO dissociation due to reduced back bonding from the rhodium into the antibonding orbitals of the CO.Ā In going from the chloride to the acetate catalyst an effect was indeed observed (70 % selectivity to oxygenates cf. 45 %). This may be tentatively correlated to the TPR profilesĀ (Figures 1 and 2) where it can be seen that the chloride prepared catalyst was indeed harder to reduce than the acetate catalyst.
The reduced activity of the acetate prepared catalyst may be explained by the alloying, seen on reduction, between the additives and the Rh as evidenced by the XRD. Direct bonding between the active Rh metal and the additives could result in the loss of active centres, thought to arise from the close proximity of the individual catalyst components. The proposed alloy formation was further investigated using temperature programmed reduction followed by XRD (Figure 3), this showed that the alloy formation did not occur until temperatures above 330 oC. Indeed reduction of the acetate catalyst at 330 oC, a temperature at which no alloy formation was observed, gave enhanced conversion (4%) and oxygenate selectivity (55%), although this was still significantly less active than the chloride prepared catalyst.
A temperature programmed oxidation of the used catalysts, followed by TGA/MS was used to investigate the nature of the carbon lay down for the two systems. Indeed the results of this set of experiments further exemplified the differences between the two precursors. Two catalysts, run on stream for similar lengths of time, were subject to a temperature ramp in a flow of 2% oxygen/argon. Over the course of the ramp the exit gas was monitored by mass spectrometry and the mass change of the catalyst monitored via TGA. The results of this set of experiments are shown in Figures 4 and 5. The masses followed were water (18), CO (28) and CO2 (44), this experiment was performed to give an idea of the amount/type of coke formed during the reaction on the two catalysts. The results clearly showed that the less active, acetate prepared catalyst (Figure 5) lost most of the carbon deposit at about 225 oC although the catalyst continued to lose mass up to 600 oC. The chloride prepared catalyst (Figure 4) however showed completely different behaviour with the first significant mass loss not occurring until 300 oC. This was followed by additional mass losses at 400 and 575 oC.
Similar experiments were also carried out in a hydrogen/nitrogen mix rather than an oxygen mix to further investigate the nature of the residual carbon species. Initially methane, acetate, benzene and napthalene ion fragments were looked for. Although the sample clearly lost mass no change in signal was observed for any of the fragments. A repeat experiment where mass fragments associated with CO and CO2 gave positive results. This suggested that the residual carbon on the surface was present either as strongly adsorbed CO or as carbonate rather than a carbonaceous residue. To further clarify this situation the experiment was repeated in Ar alone. Again only CO and CO2 were detected. After the temperature ramp in Ar a TPO was carried out on the same sample. No additional weight loss was observed further evidencing the fact that the only carbon species on the surface after reaction were either CO or carbonates. Conclusions
ĀThe implications of this set of experiments are that not only does the nature of the catalyst precursor affect the activity and the particle size and morphology of the catalyst, effects which could be expected but also the nature of the carbon species on the surface. The exact nature of the carbon on the surface is difficult to determine but all evidence points to the existence of CO and carbonates rather than carbon lay down.
References
[1] Orita, H., S. Naito, et al. (1984). J. Catal. 90(2): 183-193.
[2] Nakajo, T., K. Sano, et al. (1986). Chem. Lett. (9): 1557-1560.
[3] Nakajo, T., K. Sano, et al. (1987). J.Chem.l Soc.-Chemical Communications(9): 647-649.
[4] Yin, H. M., Y. J. Ding, et al. (2003). Energy & Fuels 17(6): 1401-1406.
[5] Luo, H. Y., W. Zhang, et al. (2001). Appl. Cat. a-Gen. 214(2): 161-166. Figures 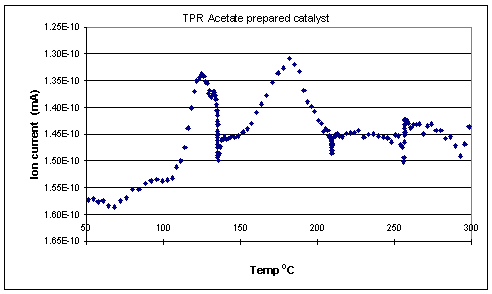
Figure 1. TPR profile of fresh catalyst prepared from rhodium chloride. 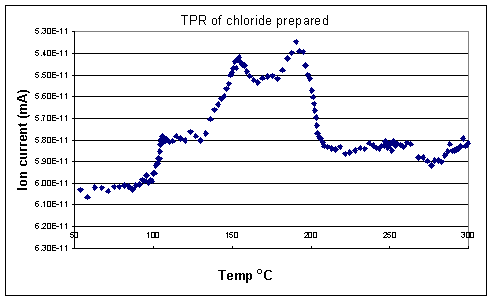
Figure 2. TPR profile of fresh catalyst prepared from rhodium acetate.
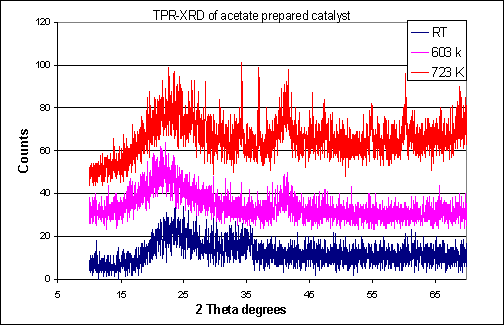
Figure 3. TPR-XRD ofĀ catalyst prepared from rhodium acetate.
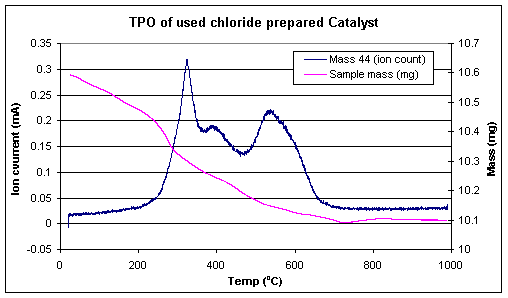
Figure 4. TPO profile of used catalyst prepared from rhodium chloride.
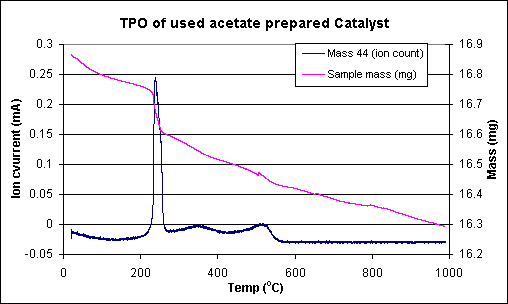
Figure 5. TPR profile of used catalyst prepared from rhodium chloride.