244b Adsorption from Dilute Liquid Solutions: Molecular Dynamics Simulations Using the Mean Force Method
The problem with direct molecular dynamics simulations for the adsorption from dilute solutions is the strong scattering of the local density of the solute [1]. In order to overcome this problem we suggested [1] to use the concept of the mean force (MF). Calculate the MF on a particle in a fixed position and then integrate the mean force along some path from r1 to r2 to obtain the difference in the potential of mean force Δw = w(r2) - w(r1) which is related to the ratio of the local density values n(r) of the solute at the initial and the final point of the integration path according to
n(r2)/n(r1) = exp(-βΔw). (1)
The method has also been applied to study the adsorption of benzene from an aqueous solution [2].
Here, we want to address two items. The first is the determination of the absolute value of the local density from the MF method and the second is the application of the MF method to study adsorption of pentaglycine.
(1) As stated above, the MF-method gives in first instant only a ratio of local densities and the question is how to determine one absolute value, e.g. n(r1). Note, that these absolute values are required for the determination of the surface excess amount and, hence, for the determination of the adsorption isotherm. In order to obtain that absolute value, we use the particle balance, which says that the integral over the local density must yield the total number of solute particles N. Starting from Eq. (1) we readily obtain
∫n(r2) dr2 = ∫n(r1) exp(-βΔw) dr2 = N, (2)
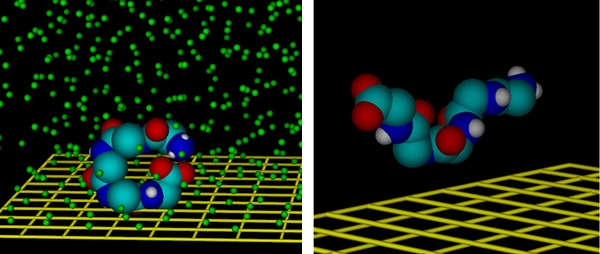
which allows the calculation of n(r1). The MF method including the particle balance has been tested in comparison with the usual direct MD simulations for the adsorption of ethane from a solution in liquid methane on a graphite surface. We found that at higher concentrations both methods yield nearly the same absolute values for the local density and, hence, yield the same adsorption isotherm values. At low concentrations and in particular in the Henry regime, however, the MF method is superior over the direct simulations. (2) In order to proceed to more complicated molecules we studied the adsorption of pentaglycine on graphite from a solution in a nonpolar solvent (argon) and from a solution in water with the MF method. Results for the structures and orientations of the pentaglycine will be shown. In order to specify the orientations we calculated the moment of inertia tensor. The figure shows in (a) that pentaglycine forms a ring in the nonpolar solvent, and in (b) that it becomes stretched in water (whilst the water molecules are not shown in the figure they have been included in the simulation). In the latter case we observed also a certain tendency for folding. Finally, we acknowledge that all simulations have been made with the code MACSIMUS [3].
[1] W. Billles, F. Bazant-Hegemark, M. Mecke, M. Wendland, and J. Fischer, Langmuir 19, 10862 (2003).
[2] R. Tscheliessnig, W. Billes, M. Wendland, J. Fischer, and J. Kolafa, Molec. Simul. 31, 661 (2005).
[3] J. Kolafa, http://www.vscht.cz/fch/software/macsimus/index.html