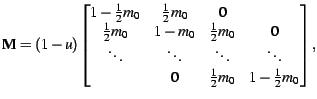> M <- steppingstone(c(.1,.2),10)generates a migration matrix with
> M
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 0.81 0.09 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
[2,] 0.09 0.72 0.09 0.00 0.00 0.00 0.00 0.00 0.00 0.00
[3,] 0.00 0.09 0.72 0.09 0.00 0.00 0.00 0.00 0.00 0.00
[4,] 0.00 0.00 0.09 0.72 0.09 0.00 0.00 0.00 0.00 0.00
[5,] 0.00 0.00 0.00 0.09 0.72 0.09 0.00 0.00 0.00 0.00
[6,] 0.00 0.00 0.00 0.00 0.09 0.72 0.09 0.00 0.00 0.00
[7,] 0.00 0.00 0.00 0.00 0.00 0.09 0.72 0.09 0.00 0.00
[8,] 0.00 0.00 0.00 0.00 0.00 0.00 0.09 0.72 0.09 0.00
[9,] 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.09 0.72 0.09
[10,] 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.09 0.81
In addition, let's assume that each subpopulation has
effective size ![]() individuals. This is done by
creating a vector Ne with elements specifying the
effective size of each subpopulation:
individuals. This is done by
creating a vector Ne with elements specifying the
effective size of each subpopulation:
> Ne <- rep(100,10)
> Ne
[1] 100 100 100 100 100 100 100 100 100 100
We can now create some data by stochastic simulation using the simulate function. Required arguments are a matrix specifying the migration rates and the vector specifying effective populations sizes. If no additional optional arguments are given, a single vector of gene frequencies is returned:
> simulate(M,Ne)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 0.285 0.315 0.44 0.61 0.655 0.495 0.495 0.46 0.575 0.425
M and Ne must, of course, have matching
dimensions. To simulate data from more than one locus, the
optional argument nloci should be given. A matrix of
gene frequencies is then returned, where each row represent
different loci. In this example, the returned matrix is
stored in p:
> p <- simulate(M,Ne,nloci=10)
> p
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 0.520 0.455 0.505 0.640 0.730 0.715 0.455 0.560 0.630 0.515
[2,] 0.635 0.535 0.485 0.530 0.610 0.450 0.370 0.415 0.515 0.510
[3,] 0.535 0.375 0.295 0.355 0.475 0.570 0.715 0.610 0.665 0.660
[4,] 0.615 0.555 0.535 0.550 0.485 0.565 0.470 0.475 0.325 0.465
[5,] 0.390 0.440 0.445 0.435 0.550 0.510 0.385 0.385 0.610 0.650
[6,] 0.705 0.580 0.490 0.410 0.565 0.480 0.565 0.745 0.750 0.755
[7,] 0.445 0.365 0.340 0.475 0.575 0.670 0.640 0.635 0.580 0.585
[8,] 0.490 0.395 0.545 0.395 0.310 0.320 0.390 0.325 0.280 0.250
[9,] 0.705 0.625 0.570 0.610 0.410 0.460 0.395 0.335 0.245 0.145
[10,] 0.320 0.300 0.400 0.255 0.320 0.450 0.440 0.425 0.495 0.295
In real life, one would typically load the gene frequency
data matrix into R (or S-Plus) by doing something like:
> p <- matrix(scan("data.txt"),byrow=T,nrow=10)
Using the approximation of the likelihood given in the appendix of Tufto et al. (1998), the maximum likelihood estimates of the parameters, for our simulated data set, can be found using the fitmodel function. This function takes two arguments; the name FUN of the migration model, and the gene frequency matrix:
> fit1 <- fitmodel(steppingstone,p)(The above call consumes about 33seconds of cpu-time on the SGI Indy unix box I have been using. With R running under Linux on a 90MHz Pentium processor it consumes about 25seconds. )
The $parameter component of the returned list contains the maximum likelihood estimates:
> fit1$par [1] 0.004024386 0.128945698and the $objective component contains the maximum likelihood:
> fit1$obj [1] 125.4102
As illustrated by this example, the estimates obtained are, as they should, not excessivly far from the true values. If you sit down in front of your computer and actually do the above, you will of course get slightly different answers, which should come as any surprise, given that the data is random.
The fitmodel function is only a simple interface to, depending on whether you use R or S-plus, S-Plus' non-linear optimizer nlminb or R's nlm function. The list returned by nlminb or nlm is in both cases slightly modified so that the $objective component contains the maximum log likelihood and the parameter component contains the maximum likelihood estimates.